
Please sign in so that we can notify you about a reply
Indications/Uses
Hypercholesterolemia: An adjunct to diet for reduction of elevated total cholesterol, LDL cholesterol, apolipoprotein B and triglycerides in patients with primary hypercholesterolemia including familial hypercholesterolemia (heterozygous variant) or combined (mixed) hyperlipidemia (corresponding to types IIa and IIb of the Fredrickson classification) when response to diet and other nonpharmacological measures is inadequate. Atorvastatin is also indicated to reduce total-C and LDL-C in patients with homozygous familial hypercholesterolemia as an adjunct to other lipid-lowering treatments (eg, LDL apheresis) or if such treatments are unavailable.
Prevention of Cardiovascular Disease: In patients estimated to have a high risk for a 1st cardiovascular event (see Pharmacology: Pharmacodynamics under Actions), as an adjunct to correction of other risk factors.
Prevention of Cardiovascular Disease: In patients estimated to have a high risk for a 1st cardiovascular event (see Pharmacology: Pharmacodynamics under Actions), as an adjunct to correction of other risk factors.
Dosage/Direction for Use
The patient should be placed on a standard cholesterol-lowering diet before receiving atorvastatin and should continue on this diet during treatment with atorvastatin. Dosage should be individualized according to baseline LDL-C levels, the goal of therapy and patient response.
Usual Starting Dose: 10 mg once a day. Adjustment of dosage should be made at intervals of ≥4 weeks. Maximum Dose: 80 mg once a day.
Current consensus guidelines should be consulted to establish treatment goals for individual patients.
Primary Hypercholesterolemia and Combined (Mixed) Hyperlipidemia: The majority of patients are controlled with atorvastatin 10 mg once a day. A therapeutic response is evident within 2 weeks and the maximum therapeutic response is usually achieved within 4 weeks. The response is maintained during chronic therapy.
Heterozygous Familial Hypercholesterolemia: Initially 10 mg daily. Doses should be individualized and adjusted every 4 weeks to 40 mg daily. Thereafter, either the dose may be increased to a maximum of 80 mg daily or a bile acid sequestrant may be combined with atorvastatin 40 mg once daily.
Homozygous Familial Hypercholesterolemia: 10-80 mg daily.
In a compassionate-use study of 64 patients, there were 46 patients for whom confirmed LDL receptor information was available. From these 46 patients, the mean percent reduction in LDL-C was approximately 21%. Atorvastatin was administered at doses up to 80 mg/day.
Atorvastatin should be used as an adjunct to other lipid-lowering treatments (eg, LDL apheresis) in these patients or if such treatments are unavailable.
Prevention of Cardiovascular Disease: In the primary prevention trials, the dose was 10 mg/day. Higher dosages may be necessary in order to attain (LDL-) cholesterol levels according to current guidelines.
Children and Adolescents: Recommended Starting Dose: 10 mg/day. The dose may be increased to 80 mg daily, according to the response and tolerability.
Pediatric use should only be carried out by specialists. Experience in pediatrics is limited to a small number of patients (4-17 years) with severe dyslipidemias eg, homozygous familial hypercholesterolemia (see Pharmacology: Pharmacodynamics under Actions). Developmental safety data in this population have not been evaluated.
Doses >20 mg/day have not been investigated in patients aged <18 years.
Elderly: Efficacy and safety in patients >70 years using recommended doses is similar to that seen in the general population.
Renal Impairment: Renal disease has no influence on the atorvastatin plasma concentrations nor lipid effects of atorvastatin; thus, no adjustment of dose is required.
Hepatic Impairment: Atorvastatin should be used with caution in patients with hepatic impairment (see Pharmacology: Pharmacokinetics under Actions and Precautions). Atorvastatin is contraindicated in patients with active liver disease (see Contraindications).
Administration: For oral administration.
Each daily dose of atorvastatin is given all at once and may be given at any time of day with or without food.
Usual Starting Dose: 10 mg once a day. Adjustment of dosage should be made at intervals of ≥4 weeks. Maximum Dose: 80 mg once a day.
Current consensus guidelines should be consulted to establish treatment goals for individual patients.
Primary Hypercholesterolemia and Combined (Mixed) Hyperlipidemia: The majority of patients are controlled with atorvastatin 10 mg once a day. A therapeutic response is evident within 2 weeks and the maximum therapeutic response is usually achieved within 4 weeks. The response is maintained during chronic therapy.
Heterozygous Familial Hypercholesterolemia: Initially 10 mg daily. Doses should be individualized and adjusted every 4 weeks to 40 mg daily. Thereafter, either the dose may be increased to a maximum of 80 mg daily or a bile acid sequestrant may be combined with atorvastatin 40 mg once daily.
Homozygous Familial Hypercholesterolemia: 10-80 mg daily.
In a compassionate-use study of 64 patients, there were 46 patients for whom confirmed LDL receptor information was available. From these 46 patients, the mean percent reduction in LDL-C was approximately 21%. Atorvastatin was administered at doses up to 80 mg/day.
Atorvastatin should be used as an adjunct to other lipid-lowering treatments (eg, LDL apheresis) in these patients or if such treatments are unavailable.
Prevention of Cardiovascular Disease: In the primary prevention trials, the dose was 10 mg/day. Higher dosages may be necessary in order to attain (LDL-) cholesterol levels according to current guidelines.
Children and Adolescents: Recommended Starting Dose: 10 mg/day. The dose may be increased to 80 mg daily, according to the response and tolerability.
Pediatric use should only be carried out by specialists. Experience in pediatrics is limited to a small number of patients (4-17 years) with severe dyslipidemias eg, homozygous familial hypercholesterolemia (see Pharmacology: Pharmacodynamics under Actions). Developmental safety data in this population have not been evaluated.
Doses >20 mg/day have not been investigated in patients aged <18 years.
Elderly: Efficacy and safety in patients >70 years using recommended doses is similar to that seen in the general population.
Renal Impairment: Renal disease has no influence on the atorvastatin plasma concentrations nor lipid effects of atorvastatin; thus, no adjustment of dose is required.
Hepatic Impairment: Atorvastatin should be used with caution in patients with hepatic impairment (see Pharmacology: Pharmacokinetics under Actions and Precautions). Atorvastatin is contraindicated in patients with active liver disease (see Contraindications).
Administration: For oral administration.
Each daily dose of atorvastatin is given all at once and may be given at any time of day with or without food.
Overdosage
There is no specific treatment for atorvastatin overdosage. In the event of an overdosage, the patient should be treated symptomatically and supportive measures instituted as required to ensure the functioning of vital organs. Due to extensive drug-binding to plasma proteins, hemodialysis is not expected to significantly enhance atorvastatin elimination from the body.
Administration
May be taken with or without food.
Contraindications
Hypersensitivity to atorvastatin calcium or to any components of Avamax.
Active liver disease or unexplained persistent elevations of serum transaminases and diseases of skeletal muscles.
Use in pregnancy & lactation: Atorvastatin is contraindicated during pregnancy and breastfeeding. Cholesterol and other substances, which are formed during its biosynthesis, are essential for fetus development (also formation of steroids and cell membranes). 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors reduce cholesterol synthesis and possibly also synthesis of other biologically active substances which are cholesterol derivatives; its administration in pregnancy may therefore induce harmful effects in fetus. Usage of HMG-CoA reductase inhibitors is thus contraindicated in pregnancy and breastfeeding. Atorvastatin may be administered to women of childbearing potential only when such patients are highly unlikely to conceive and have been informed of the potential harmful effects of Avamax. If the woman becomes pregnant while taking atorvastatin, it should be discontinued and the patient advised again as to the potential hazards to the fetus.
Because of the potential for adverse reactions in nursing infants, women taking atorvastatin should not breastfeed.
Active liver disease or unexplained persistent elevations of serum transaminases and diseases of skeletal muscles.
Use in pregnancy & lactation: Atorvastatin is contraindicated during pregnancy and breastfeeding. Cholesterol and other substances, which are formed during its biosynthesis, are essential for fetus development (also formation of steroids and cell membranes). 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors reduce cholesterol synthesis and possibly also synthesis of other biologically active substances which are cholesterol derivatives; its administration in pregnancy may therefore induce harmful effects in fetus. Usage of HMG-CoA reductase inhibitors is thus contraindicated in pregnancy and breastfeeding. Atorvastatin may be administered to women of childbearing potential only when such patients are highly unlikely to conceive and have been informed of the potential harmful effects of Avamax. If the woman becomes pregnant while taking atorvastatin, it should be discontinued and the patient advised again as to the potential hazards to the fetus.
Because of the potential for adverse reactions in nursing infants, women taking atorvastatin should not breastfeed.
Special Precautions
Liver Dysfunction: Usage of HMG-CoA reductase inhibitors eg, administration of some other lipid-lowering agents, has been associated with biochemical abnormalities of liver function.
It is recommended that liver function tests be performed prior to treatment and after 12 weeks following the initiation of therapy or at any elevation of dose and periodically, eg, semi-annually, thereafter.
Changes of liver enzymes generally occur in the first 3 months of treatment with atorvastatin. Patients who develop increased transaminase levels should be monitored until the abnormalities resolve. Should an increase in alanine aminotransferase (ALT) or aspartate aminotransferase (AST) value exceed 3 times the upper limit value, reduction of dose or discontinuation of atorvastatin treatment is recommended. Atorvastatin should be used with great caution in patients who consume substantial quantities of alcohol and/or have a history of liver disease. Use of atorvastatin in patients with active liver disease or unexplained persistent transaminase elevations is contraindicated.
Skeletal Muscles: The possibility of myopathy [muscle aches or muscle weakness in conjunction with increases in creatine phosphokinase (CPK) values >10 times the upper limit value] should be considered in any patient with diffuse myalgia, muscle tenderness or weakness, and/or marked elevation of CPK. The risk of myopathy in patients taking statins is increased with concurrent administration of cyclosporine, fibric acid derivatives, erythromycin, immunosuppressive drugs, azole antifungals or lipid-lowering doses of niacin should carefully weigh the potential benefits and risks, and should carefully monitor all patients who develop the signs or symptoms of muscle pain, tenderness or weakness, particularly during the initial months of therapy and during any periods of upward dosage titration of either medicinal product.
Atorvastatin therapy should be temporarily withheld or discontinued in any patient with an acute, serious condition which might result from a myopathy and in those having a risk factor predisposing to the development of renal failure secondary to rhabdomyolysis (eg, severe acute infection, hypotension, major surgery, trauma, severe metabolic, endocrine and electrolyte disorders and uncontrolled seizures).
Effects on the Ability to Drive or Operate Machinery: Atorvastatin does not affect the ability to drive motor vehicles and use machines.
It is recommended that liver function tests be performed prior to treatment and after 12 weeks following the initiation of therapy or at any elevation of dose and periodically, eg, semi-annually, thereafter.
Changes of liver enzymes generally occur in the first 3 months of treatment with atorvastatin. Patients who develop increased transaminase levels should be monitored until the abnormalities resolve. Should an increase in alanine aminotransferase (ALT) or aspartate aminotransferase (AST) value exceed 3 times the upper limit value, reduction of dose or discontinuation of atorvastatin treatment is recommended. Atorvastatin should be used with great caution in patients who consume substantial quantities of alcohol and/or have a history of liver disease. Use of atorvastatin in patients with active liver disease or unexplained persistent transaminase elevations is contraindicated.
Skeletal Muscles: The possibility of myopathy [muscle aches or muscle weakness in conjunction with increases in creatine phosphokinase (CPK) values >10 times the upper limit value] should be considered in any patient with diffuse myalgia, muscle tenderness or weakness, and/or marked elevation of CPK. The risk of myopathy in patients taking statins is increased with concurrent administration of cyclosporine, fibric acid derivatives, erythromycin, immunosuppressive drugs, azole antifungals or lipid-lowering doses of niacin should carefully weigh the potential benefits and risks, and should carefully monitor all patients who develop the signs or symptoms of muscle pain, tenderness or weakness, particularly during the initial months of therapy and during any periods of upward dosage titration of either medicinal product.
Atorvastatin therapy should be temporarily withheld or discontinued in any patient with an acute, serious condition which might result from a myopathy and in those having a risk factor predisposing to the development of renal failure secondary to rhabdomyolysis (eg, severe acute infection, hypotension, major surgery, trauma, severe metabolic, endocrine and electrolyte disorders and uncontrolled seizures).
Effects on the Ability to Drive or Operate Machinery: Atorvastatin does not affect the ability to drive motor vehicles and use machines.
Use In Pregnancy & Lactation
Use in pregnancy & lactation: Atorvastatin is contraindicated during pregnancy and breastfeeding. Cholesterol and other substances, which are formed during its biosynthesis, are essential for fetus development (also formation of steroids and cell membranes). 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors reduce cholesterol synthesis and possibly also synthesis of other biologically active substances which are cholesterol derivatives; its administration in pregnancy may therefore induce harmful effects in fetus. Usage of HMG-CoA reductase inhibitors is thus contraindicated in pregnancy and breastfeeding. Atorvastatin may be administered to women of childbearing potential only when such patients are highly unlikely to conceive and have been informed of the potential harmful effects of Avamax. If the woman becomes pregnant while taking atorvastatin, it should be discontinued and the patient advised again as to the potential hazards to the fetus.
Because of the potential for adverse reactions in nursing infants, women taking atorvastatin should not breastfeed.
Because of the potential for adverse reactions in nursing infants, women taking atorvastatin should not breastfeed.
Adverse Reactions
Atorvastatin is generally well tolerated. Adverse effects are usually mild and transient. In >1% of patients administered atorvastatin, constipation, flatulence, dyspepsia, abdominal pain, headache, nausea, muscle aches, diarrhea and insomnia were observed.
As follows is a report of the adverse events, regardless of the cause of their appearance, which have developed in patients treated with atorvastatin in clinical trials. The events occurred in ≥2% and <2% of patients.
Body as a Whole: Chest pain, face edema, fever, neck rigidity, malaise, photosensitivity reaction, generalized edema.
Digestive System: Nausea, gastroenteritis, abnormal values of liver function tests, colitis, vomiting, gastritis, dry mouth, rectal hemorrhage, esophagitis, eructation, glossitis, mouth erosion, anorexia, increased appetite, stomatitis, biliary pain, cheilitis, duodenal ulcer, dysphagia, enteritis, melena, gum hemorrhage, stomach ulcer, tenesmus, ulcerative stomatitis, hepatitis, pancreatitis, cholestatic jaundice.
Respiratory System: Bronchitis, rhinitis, pneumonia, dyspnea, asthma, epistaxis.
Nervous System: Insomnia, dizziness, paresthesia, somnolence, amnesia, abnormal dreams, decreased libido, emotional lability, incoordination, peripheral neuropathy, torticollis, facial paralysis, hyperkinesia, depression, hypesthesia, hypertonia.
Musculoskeletal System: Arthritis, leg cramps, bursitis, tenosynovitis, myasthenia, tendinous contracture, myositis.
Skin: Pruritus, contact dermatitis, alopecia, dry skin, sweating, acne, urticaria, eczema, seborrhea, skin ulcer.
Urogenital System: Urinary tract infection, urinary frequency, cystitis, hematuria, impotence, dysuria, kidney calculus, nocturia, epididymitis, fibrocystic breast, vaginal hemorrhage, albuminuria, breast enlargement, metorrhagia, nephritis, urinary incontinence, urinary retention, urinary urgency, abnormal ejaculation, uterine hemorrhage.
Special Senses: Amblyopia, tinnitus, dry eyes, refraction disorder, eye hemorrhage, deafness, glaucoma, parosmia, taste loss, taste perversion.
Cardiovascular System: Palpitation, vasodilation, syncope, migraine, postural hypotension, phlebitis, arrhythmia, angina pectoris, hypertension.
Metabolic and Nutritional Disorders: Peripheral edema, hyperglycemia, increased creatine phosphokinase, gout, body weight gain, hypoglycemia.
Hemic and Lymphatic System: Ecchymosis, anemia, lymphadenopathy, thrombocytopenia, petechia.
As follows is a report of the adverse events, regardless of the cause of their appearance, which have developed in patients treated with atorvastatin in clinical trials. The events occurred in ≥2% and <2% of patients.
Body as a Whole: Chest pain, face edema, fever, neck rigidity, malaise, photosensitivity reaction, generalized edema.
Digestive System: Nausea, gastroenteritis, abnormal values of liver function tests, colitis, vomiting, gastritis, dry mouth, rectal hemorrhage, esophagitis, eructation, glossitis, mouth erosion, anorexia, increased appetite, stomatitis, biliary pain, cheilitis, duodenal ulcer, dysphagia, enteritis, melena, gum hemorrhage, stomach ulcer, tenesmus, ulcerative stomatitis, hepatitis, pancreatitis, cholestatic jaundice.
Respiratory System: Bronchitis, rhinitis, pneumonia, dyspnea, asthma, epistaxis.
Nervous System: Insomnia, dizziness, paresthesia, somnolence, amnesia, abnormal dreams, decreased libido, emotional lability, incoordination, peripheral neuropathy, torticollis, facial paralysis, hyperkinesia, depression, hypesthesia, hypertonia.
Musculoskeletal System: Arthritis, leg cramps, bursitis, tenosynovitis, myasthenia, tendinous contracture, myositis.
Skin: Pruritus, contact dermatitis, alopecia, dry skin, sweating, acne, urticaria, eczema, seborrhea, skin ulcer.
Urogenital System: Urinary tract infection, urinary frequency, cystitis, hematuria, impotence, dysuria, kidney calculus, nocturia, epididymitis, fibrocystic breast, vaginal hemorrhage, albuminuria, breast enlargement, metorrhagia, nephritis, urinary incontinence, urinary retention, urinary urgency, abnormal ejaculation, uterine hemorrhage.
Special Senses: Amblyopia, tinnitus, dry eyes, refraction disorder, eye hemorrhage, deafness, glaucoma, parosmia, taste loss, taste perversion.
Cardiovascular System: Palpitation, vasodilation, syncope, migraine, postural hypotension, phlebitis, arrhythmia, angina pectoris, hypertension.
Metabolic and Nutritional Disorders: Peripheral edema, hyperglycemia, increased creatine phosphokinase, gout, body weight gain, hypoglycemia.
Hemic and Lymphatic System: Ecchymosis, anemia, lymphadenopathy, thrombocytopenia, petechia.
Drug Interactions
The risk of myopathy in patients taking statins is increased with concurrent administration of cyclosporine, fibric acid derivatives, erythromycin, niacin or azole antifungals.
Antacids: When atorvastatin and antacid suspension containing magnesium and aluminum hydroxide are co-administered, plasma concentration of atorvastatin decreases approximately by 35%. However, LDL cholesterol reduction is not altered.
Colestipol: Plasma concentrations of atorvastatin decreased approximately 25% when atorvastatin and colestipol were co-administered. However, reduction of LDL cholesterol was greater when atorvastatin and colestipol were co-administered than when either medicinal product was given alone.
Digoxin: When multiple doses of atorvastatin and digoxin are co-administered, a steady state plasma digoxin concentraton is increased by approximately 20%. Patients taking digoxin should be monitored appropriately.
Erythromycin: In healthy individuals, plasma concentrations of atorvastatin increased on average by 40% with co-administration of atorvastatin and erythromycin, a known inhibitor of CYP450 3A4.
Oral Contraceptives: Co-administration of atorvastatin and an oral contraceptive increased area under the curve (AUC) values for norethindrone by approximately 30% and of ethinyl estradiol by 20%, respectively. These increases should be considered when selecting a suitable oral contraceptive for a woman taking atorvastatin.
Warfarin: In patients on chronic warfarin treatment, atorvastatin slightly shortens the prothrombin time in the first days of its administration; this time is normalized, however, after 15 days of therapy. In patients receiving warfarin, the prothrombin time should be controlled more often after atorvastatin introduction.
Incompatibilities: Not applicable.
Antacids: When atorvastatin and antacid suspension containing magnesium and aluminum hydroxide are co-administered, plasma concentration of atorvastatin decreases approximately by 35%. However, LDL cholesterol reduction is not altered.
Colestipol: Plasma concentrations of atorvastatin decreased approximately 25% when atorvastatin and colestipol were co-administered. However, reduction of LDL cholesterol was greater when atorvastatin and colestipol were co-administered than when either medicinal product was given alone.
Digoxin: When multiple doses of atorvastatin and digoxin are co-administered, a steady state plasma digoxin concentraton is increased by approximately 20%. Patients taking digoxin should be monitored appropriately.
Erythromycin: In healthy individuals, plasma concentrations of atorvastatin increased on average by 40% with co-administration of atorvastatin and erythromycin, a known inhibitor of CYP450 3A4.
Oral Contraceptives: Co-administration of atorvastatin and an oral contraceptive increased area under the curve (AUC) values for norethindrone by approximately 30% and of ethinyl estradiol by 20%, respectively. These increases should be considered when selecting a suitable oral contraceptive for a woman taking atorvastatin.
Warfarin: In patients on chronic warfarin treatment, atorvastatin slightly shortens the prothrombin time in the first days of its administration; this time is normalized, however, after 15 days of therapy. In patients receiving warfarin, the prothrombin time should be controlled more often after atorvastatin introduction.
Incompatibilities: Not applicable.
Storage
Store at temperatures not exceeding 30°C.
Action
Pharmacotherapeutic Group: HMG-CoA-reductase inhibitors.
Pharmacology: Pharmacodynamics: Atorvastatin is a selective, competitive inhibitor of HMG-CoA reductase, the rate-limiting enzyme responsible for the conversion of 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) to mevalonate, a precursor of sterols, including cholesterol. Triglycerides (TG) and cholesterol in the liver are incorporated into very low-density lipoproteins (VLDL) and released into the plasma for delivery to peripheral tissues. Low density lipoprotein (LDL) is formed from VLDL and is catabolized primarily through the receptor with high affinity to LDL (LDL receptor).
Atorvastatin lowers plasma cholesterol and lipoprotein serum concentrations by inhibiting HMG-CoA reductase and subsequently cholesterol biosynthesis in the liver and increases the number of hepatic LDL receptors on the cell surface for enhanced uptake and catabolism of LDL.
Atorvastatin reduces LDL production and the number of LDL particles. Atorvastatin produces a profound and sustained increase in LDL receptor activity coupled with a beneficial change in the quality of circulating LDL particles. Atorvastatin is effective in reducing LDL-C in patients with homozygous familial hypercholesterolemia, a population that has not usually responded to lipid lowering medication.
Atorvastatin has been shown to reduce concentrations of total-C (TC) (30-46%), LDL-C (41-61%), apolipoprotein B (34-50%) and triglycerides (14-33%) while producing variable increases in high density lipoprotein (HDL)-C and apolipoprotein A1 in a dose response study.
These results are consistent in patients with heterozygous familial hypercholesterolemia, nonfamilial forms of hypercholesterolemia and mixed hyperlipidemia, including patients with noninsulin-dependent diabetes mellitus.
Reductions in total-C, LDL-C and apolipoprotein B have been proven to reduce risk for cardiovascular events and mortality.
Atherosclerosis: In the reversing atherosclerosis with aggressive lipid-lowering study (REVERSAL), the effect of intensive lipid-lowering with atorvastatin 80 mg and standard degree of lipid-lowering with pravastatin 40 mg on coronary atherosclerosis was assessed by intravascular ultrasound (IVUS), during angiography, in patients with coronary heart disease (CHD). In this randomized, double-blind, multicenter-controlled clinical trial, IVUS was performed at baseline and at 18 months in 502 patients. In the atorvastatin group (n=253), there was no progression of atherosclerosis.
The median percent change, from baseline, in total atheroma volume (the primary study criteria) was -0.4% (p=0.98) in the atorvastatin group and +2.7% (p=0.001) in the pravastatin group (n=249). When compared to pravastatin, the effects of atorvastatin were statistically significant (p=0.02). The effect of intensive lipid-lowering with atorvastatin on cardiovascular coronary death was not investigated in this study. Therefore, the clinical significance of these imaging results with regard to the primary and secondary prevention of cardiovascular events is unknown.
In the atorvastatin group, LDL-C was reduced to a mean of 2.04 mmol/L±0.8 (78.9 mg/dL±30) from baseline 3.89 mmol/L±0.7 (150 mg/dL±28) and in the pravastatin group, LDL-C was reduced to a mean of 2.85 mmol/L±0.7 (110 mg/dL±26) from baseline 3.89 mmol/L±0.7 (150 mg/dL±26) (p<0.0001). Atorvastatin also significantly reduced mean TC by 34.1% (pravastatin: -18.4%, p<0.0001), mean TG levels by 20% (pravastatin: -6.8%, p<0.0009) and mean apolipoprotein B by 39.1% (pravastatin: -22%, p<0.0001). Atorvastatin increased mean HDL-C by 2.9% (pravastatin: +5.6%, p=NS). There was a 36.4% mean reduction in CRP in the atorvastatin group compared to a 5.2% reduction in the pravastatin group (p<0.0001).
Study results were obtained with the 80 mg dose strength. Therefore, they cannot be extrapolated to the lower dose strengths.
The safety and tolerability profiles of the 2 treatment groups were comparable.
Heterozygous Familial Hypercholesterolemia in Pediatric Patients: In a double-blind, placebo-controlled study followed by an open-label phase, 187 boys and postmenarchal girls 10-17 years (mean age 14.1 years) with heterozygous familial hypercholesterolemia (FH) or severe hypercholesterolemia were randomized to atorvastatin (n=140) or placebo (n=47) for 26 weeks and then all received atorvastatin for 26 weeks. Inclusion in the study required: (1) a baseline LDL-C level 34.91 mmol/L or (2) a baseline LDL-C 34.14 mmol/L and positive family history of FH or documented premature cardiovascular disease in a 1st- or 2nd-degree relative. The mean baseline LDL-C value was 5.65 mmol/L (range: 3.58-9.96 mmol/L) in the atorvastatin group compared to 5.95 mmol/L (range: 4.14-8.39 mmol/L) in placebo group. The dosage of atorvastatin (once daily) was 10 mg for the first 4 weeks and up-titrated to 20 mg if the LDL-C level was >3.36 mmol/L. The number of atorvastatin-treated patients who required up-titration to 20 mg after week 4 during the double-blind phase was 80 (57.1%).
Atorvastatin significantly decreased plasma levels of total-C, LDL-C, triglycerides and apolipoprotein B (Apo) during the 26 week double-blind phase. (See Table 1.)
Pharmacology: Pharmacodynamics: Atorvastatin is a selective, competitive inhibitor of HMG-CoA reductase, the rate-limiting enzyme responsible for the conversion of 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) to mevalonate, a precursor of sterols, including cholesterol. Triglycerides (TG) and cholesterol in the liver are incorporated into very low-density lipoproteins (VLDL) and released into the plasma for delivery to peripheral tissues. Low density lipoprotein (LDL) is formed from VLDL and is catabolized primarily through the receptor with high affinity to LDL (LDL receptor).
Atorvastatin lowers plasma cholesterol and lipoprotein serum concentrations by inhibiting HMG-CoA reductase and subsequently cholesterol biosynthesis in the liver and increases the number of hepatic LDL receptors on the cell surface for enhanced uptake and catabolism of LDL.
Atorvastatin reduces LDL production and the number of LDL particles. Atorvastatin produces a profound and sustained increase in LDL receptor activity coupled with a beneficial change in the quality of circulating LDL particles. Atorvastatin is effective in reducing LDL-C in patients with homozygous familial hypercholesterolemia, a population that has not usually responded to lipid lowering medication.
Atorvastatin has been shown to reduce concentrations of total-C (TC) (30-46%), LDL-C (41-61%), apolipoprotein B (34-50%) and triglycerides (14-33%) while producing variable increases in high density lipoprotein (HDL)-C and apolipoprotein A1 in a dose response study.
These results are consistent in patients with heterozygous familial hypercholesterolemia, nonfamilial forms of hypercholesterolemia and mixed hyperlipidemia, including patients with noninsulin-dependent diabetes mellitus.
Reductions in total-C, LDL-C and apolipoprotein B have been proven to reduce risk for cardiovascular events and mortality.
Atherosclerosis: In the reversing atherosclerosis with aggressive lipid-lowering study (REVERSAL), the effect of intensive lipid-lowering with atorvastatin 80 mg and standard degree of lipid-lowering with pravastatin 40 mg on coronary atherosclerosis was assessed by intravascular ultrasound (IVUS), during angiography, in patients with coronary heart disease (CHD). In this randomized, double-blind, multicenter-controlled clinical trial, IVUS was performed at baseline and at 18 months in 502 patients. In the atorvastatin group (n=253), there was no progression of atherosclerosis.
The median percent change, from baseline, in total atheroma volume (the primary study criteria) was -0.4% (p=0.98) in the atorvastatin group and +2.7% (p=0.001) in the pravastatin group (n=249). When compared to pravastatin, the effects of atorvastatin were statistically significant (p=0.02). The effect of intensive lipid-lowering with atorvastatin on cardiovascular coronary death was not investigated in this study. Therefore, the clinical significance of these imaging results with regard to the primary and secondary prevention of cardiovascular events is unknown.
In the atorvastatin group, LDL-C was reduced to a mean of 2.04 mmol/L±0.8 (78.9 mg/dL±30) from baseline 3.89 mmol/L±0.7 (150 mg/dL±28) and in the pravastatin group, LDL-C was reduced to a mean of 2.85 mmol/L±0.7 (110 mg/dL±26) from baseline 3.89 mmol/L±0.7 (150 mg/dL±26) (p<0.0001). Atorvastatin also significantly reduced mean TC by 34.1% (pravastatin: -18.4%, p<0.0001), mean TG levels by 20% (pravastatin: -6.8%, p<0.0009) and mean apolipoprotein B by 39.1% (pravastatin: -22%, p<0.0001). Atorvastatin increased mean HDL-C by 2.9% (pravastatin: +5.6%, p=NS). There was a 36.4% mean reduction in CRP in the atorvastatin group compared to a 5.2% reduction in the pravastatin group (p<0.0001).
Study results were obtained with the 80 mg dose strength. Therefore, they cannot be extrapolated to the lower dose strengths.
The safety and tolerability profiles of the 2 treatment groups were comparable.
Heterozygous Familial Hypercholesterolemia in Pediatric Patients: In a double-blind, placebo-controlled study followed by an open-label phase, 187 boys and postmenarchal girls 10-17 years (mean age 14.1 years) with heterozygous familial hypercholesterolemia (FH) or severe hypercholesterolemia were randomized to atorvastatin (n=140) or placebo (n=47) for 26 weeks and then all received atorvastatin for 26 weeks. Inclusion in the study required: (1) a baseline LDL-C level 34.91 mmol/L or (2) a baseline LDL-C 34.14 mmol/L and positive family history of FH or documented premature cardiovascular disease in a 1st- or 2nd-degree relative. The mean baseline LDL-C value was 5.65 mmol/L (range: 3.58-9.96 mmol/L) in the atorvastatin group compared to 5.95 mmol/L (range: 4.14-8.39 mmol/L) in placebo group. The dosage of atorvastatin (once daily) was 10 mg for the first 4 weeks and up-titrated to 20 mg if the LDL-C level was >3.36 mmol/L. The number of atorvastatin-treated patients who required up-titration to 20 mg after week 4 during the double-blind phase was 80 (57.1%).
Atorvastatin significantly decreased plasma levels of total-C, LDL-C, triglycerides and apolipoprotein B (Apo) during the 26 week double-blind phase. (See Table 1.)

The mean achieved LDL-C value was 3.38 mmol/L (range: 1.81-6.26 mmol/L) in the atorvastatin group compared to 5.91 mmol/L (range: 3.93-9.96 mmol/L) in the placebo group during the 26-week double-blind phase.
In this limited controlled study, there was no detectable effect on growth or sexual maturation in boys or on menstrual length in girls. Atorvastatin has not been studied in controlled clinical trials involving pre-pubertal patients or patients <10 years. The safety and efficacy of doses >20 mg have not been studied in controlled trials in children. The long-term efficacy of atorvastatin therapy in childhood to reduce morbidity and mortality in adulthood has not been established.
Prevention of Cardiovascular Disease: The effect of atorvastatin on fatal and nonfatal CHD was assessed in a randomized, double-blind, placebo-controlled study, the Anglo-Scandinavian cardiac outcomes trial lipid-lowering arm (ASCOT-LLA). Patients were hypertensive, 40-79 years, with no previous myocardial infarction or treatment for angina, and with TC levels ≤6.5 mmol/L (251 mg/dL). All patients had at least 3 of the predefined cardiovascular risk factors: Male gender, ≥55 years, smoking, diabetes, history of CHD in a 1st-degree relative, TC: HDL-C >6, peripheral vascular disease, left ventricular hypertrophy, prior cerebrovascular event, specific electrocardiogram (ECG) abnormality, proteinuria/albuminuria. Not all included patients were estimated to have a high risk for a 1st cardiovascular event.
Patients were treated with antihypertensive therapy (either amlodipine or atenolol-based regimen) and either atorvastatin 10 mg daily (n=5,168) or placebo (n=5,137).
The absolute and relative risk reduction effect of atorvastatin was as follows: See Table 2.
In this limited controlled study, there was no detectable effect on growth or sexual maturation in boys or on menstrual length in girls. Atorvastatin has not been studied in controlled clinical trials involving pre-pubertal patients or patients <10 years. The safety and efficacy of doses >20 mg have not been studied in controlled trials in children. The long-term efficacy of atorvastatin therapy in childhood to reduce morbidity and mortality in adulthood has not been established.
Prevention of Cardiovascular Disease: The effect of atorvastatin on fatal and nonfatal CHD was assessed in a randomized, double-blind, placebo-controlled study, the Anglo-Scandinavian cardiac outcomes trial lipid-lowering arm (ASCOT-LLA). Patients were hypertensive, 40-79 years, with no previous myocardial infarction or treatment for angina, and with TC levels ≤6.5 mmol/L (251 mg/dL). All patients had at least 3 of the predefined cardiovascular risk factors: Male gender, ≥55 years, smoking, diabetes, history of CHD in a 1st-degree relative, TC: HDL-C >6, peripheral vascular disease, left ventricular hypertrophy, prior cerebrovascular event, specific electrocardiogram (ECG) abnormality, proteinuria/albuminuria. Not all included patients were estimated to have a high risk for a 1st cardiovascular event.
Patients were treated with antihypertensive therapy (either amlodipine or atenolol-based regimen) and either atorvastatin 10 mg daily (n=5,168) or placebo (n=5,137).
The absolute and relative risk reduction effect of atorvastatin was as follows: See Table 2.
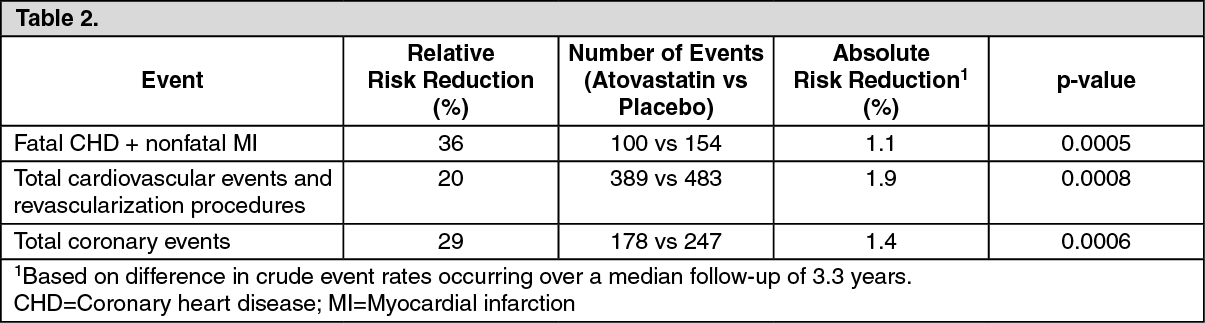
Total mortality and cardiovascular mortality were not significantly reduced (185 vs 212 events, p=0.17 and 74 vs 82 events, p=0.51). In the subgroup analyses by gender (81% males, 19% females), a beneficial effect of atorvastatin was seen in males but could not be established in females possibly due to the low event rate in the female subgroup. Overall and cardiovascular mortality were numerically higher in the female patients (38 vs 30 and 17 vs 12), but this was not statistically significant. There was significant treatment interaction by antihypertensive baseline therapy. The primary endpoint (fatal CHD plus nonfatal MI) was significantly reduced by atorvastatin in patients treated with amlodipine [Hazard Ratio (HR) 0.47 (0.32-0.69), p=0.00008], but not in those treated with atenolol [HR 0.83 (0.59-1.17), p=0.287].
The effect of atorvastatin on fatal and nonfatal cardiovascular disease was also assessed in a randomized, double-blind, multicenter, placebo-controlled trial, the collaborative atorvastatin diabetes study (CARDS) in patients with type 2 diabetes, 40-75 years, without prior history of cardiovascular disease, and with LDL-C ≤4.14 mmol/L (160 mg/dL) and TG ≤6.78 mmol/L (600 mg/dL).
All patients had at least 1 of the following risk factors: Hypertension, current smoking, retinopathy, microalbuminuria or macroalbuminuria.
Patients were treated with either atorvastatin 10 mg daily (n=1,428) or placebo (n=1,410) for a median follow-up of 3.9 years.
The absolute and relative risk reduction effect of atorvastatin was as follows: See Table 3.
The effect of atorvastatin on fatal and nonfatal cardiovascular disease was also assessed in a randomized, double-blind, multicenter, placebo-controlled trial, the collaborative atorvastatin diabetes study (CARDS) in patients with type 2 diabetes, 40-75 years, without prior history of cardiovascular disease, and with LDL-C ≤4.14 mmol/L (160 mg/dL) and TG ≤6.78 mmol/L (600 mg/dL).
All patients had at least 1 of the following risk factors: Hypertension, current smoking, retinopathy, microalbuminuria or macroalbuminuria.
Patients were treated with either atorvastatin 10 mg daily (n=1,428) or placebo (n=1,410) for a median follow-up of 3.9 years.
The absolute and relative risk reduction effect of atorvastatin was as follows: See Table 3.
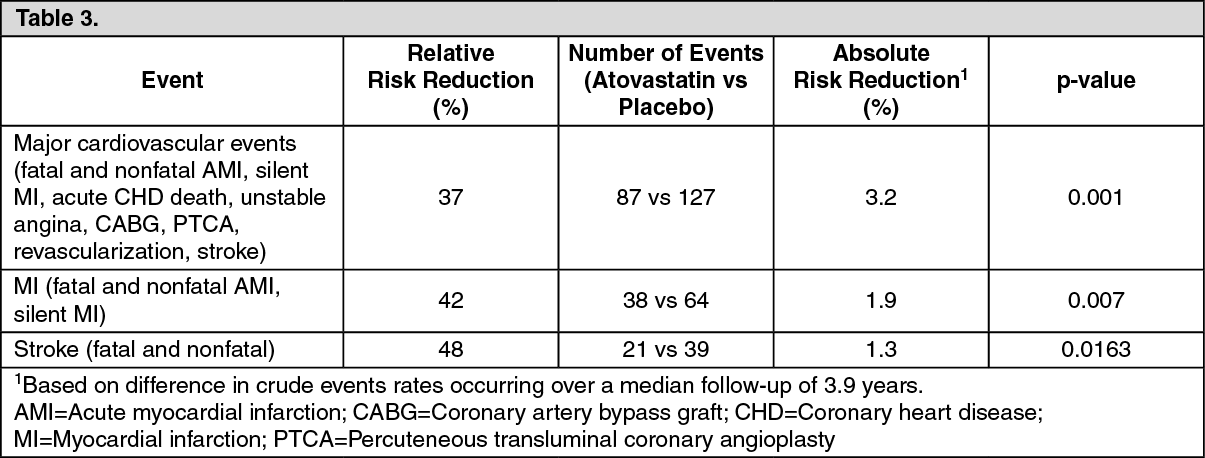
There was no evidence of a difference in the treatment effect by patient's gender, age or baseline LDL-C level. A favorable trend was observed regarding the mortality rate (82 deaths in the placebo group vs 61 deaths in the atorvastatin group, p=0.0592).
Recurrent Stroke: In the stroke prevention by aggressive reduction in cholesterol levels (SPARCL) study, the effect of atorvastatin 80 mg daily or placebo on stroke was evaluated in 4,731 patients who had a stroke or transient ischemic attack (TIA) within the preceding 6 months and no history of a CHD. Patients were male 60%, 21-92 years (average age 63 years) and had an average baseline LDL of 133 mg/dL (3.4 mmol/L). The mean LDL-C was 73 mg/dL (1.9 mmol/L) during treatment with atorvastatin and 129 mg/dL (3.3 mmol/L) during treatment with placebo. Median follow-up was 4.9 years.
Atorvastatin 80 mg reduced the risk of the primary endpoint of fatal or nonfatal stroke by 15% (HR 0.85; 95% CI, 0.72-1; p=0.05 or 0.84; 95% CI, 0.71-0.99; p=0.03 after adjustment for baseline factors) compared to placebo. All cause mortality was 9.1% (216/2,365) for atorvastatin versus 8.9% (211/2,366) for placebo.
In a post analysis, atorvastatin 80 mg reduced the incidence of ischemic stroke (218/2,365, 9.2% vs 274/2,366, 11.6%, p=0.01) and increased the incidence of hemorrhagic stroke (55/2,365, 2.3% vs 33/2,366, 1.4%, p=0.02) compared to placebo.
The risk of hemorrhagic stroke was increased in patients who entered the study with prior hemorrhagic stroke (7/45 for atorvastatin vs 2/48 for placebo; HR 4.06; 95% CI, 0.84-19.57) and the risk of ischemic stroke was similar between groups (3/45 for atorvastatin vs 2/48 for placebo; HR 1.64; 95% CI, 0.27-9.82).
The risk of hemorrhagic stroke was increased in patients who entered the study with prior lacunar infarct (20/708 for atorvastatin vs 4/701 for placebo; HR 4.99; 95% CI, 1.71-14.61), but the risk of ischemic stroke was also decreased in these patients (79/708 for atorvastatin vs 102/701 for placebo; HR 0.76; 95% CI, 0.57-1.02). It is possible that the net risk of stroke is increased in patients with prior lacunar infarct who receive atorvastatin 80 mg/day.
All cause mortality was 15.6% (7/45) for atorvastatin versus 10/4% (5/48) in the subgroup of patients with prior hemorrhagic stroke. All cause mortality was 10.9% (77/708) for atorvastatin versus 9.1% (64/701) for placebo in the subgroup of patients with prior lacunar infarct.
Pharmacokinetics: Absorption: Atorvastatin is rapidly absorbed after oral administration; maximum plasma concentrations (Cmax) occur within 1-2 hrs. Extent of absorption increases in proportion to atorvastatin dose. After oral administration, atorvastatin film-coated tablets are 95-99% bioavailable compared to the oral solution. The absolute bioavailability of atorvastatin is approximately 12% and the systemic availability of HMG-CoA reductase inhibitory activity is approximately 30%. The low systemic availability is attributed to presystemic clearance in gastrointestinal mucosa and/or hepatic first-pass metabolism.
Distribution: Mean volume of distribution of atorvastatin is approximately 381 L. Atorvastatin is ≥98% bound to plasma proteins.
Metabolism: Atorvastatin is metabolized by cytochrome P450 (CYP450) 3A4 to ortho- and parahydroxylated derivatives and various β-oxidation products. Apart from other pathways these products are further metabolized via glucuronidation. In vitro, inhibition of HMG-CoA reductase by ortho- and parahydroxylated metabolites is equivalent to that of atorvastatin. Approximately 70% of circulating inhibitory activity for HMG-CoA reductase is attributed to active metabolites.
Excretion: Atorvastatin is eliminated primarily in bile following hepatic and/or extrahepatic metabolism.
However, Avamax does not appear to undergo significant enterohepatic recirculation. Mean plasma elimination half-life (t½) of atorvastatin in humans is approximately 14 hrs. The t½ of inhibitory activity for HMG-CoA reductase is approximately 20-30 hrs due to the contribution of active metabolites.
Special Populations: Geriatric: Plasma concentrations of atorvastatin and its active metabolites are higher in healthy elderly subjects than in young adults while the lipid effects were comparable to those seen in younger patient populations.
Pediatric: Pharmacokinetic data in the pediatric population are not available.
Gender: Concentrations of atorvastatin and its active metabolites in women differ from those in men [Women: approximately 20% higher for Cmax and approximately 10% lower for area under the curve (AUC)]. These differences were of no clinical significance, resulting in no clinically significant differences in lipid effects among men and women.
Renal Insufficiency: Renal disease has no influence on the plasma concentrations or lipid effects of atorvastatin and its active metabolites.
Hepatic Insufficiency: Plasma concentrations of atorvastatin and its active metabolites are markedly increased (approximately 16-fold in Cmax and approximately 11-fold in AUC) in patients with chronic alcoholic liver disease (Childs-Pugh B).
Toxicology: Preclinical Safety Data: Atorvastatin was not carcinogenic in rats. The maximum dose used was 63-fold higher than the highest human dose (80 mg/day) on a mg/kg body-weight basis and 8- to 16-fold higher based on AUC(0-24 hr) values as determined by total inhibitory activity. In a 2-year study in mice, incidences of hepatocellular adenoma in males and hepatocellular carcinomas in females were increased at the maximum dose used, and the maximum dose used was 250-fold higher than the highest human dose on a mg/kg body-weight basis. Systemic exposure was 6- to 11-fold higher based on AUC(0-24 hr). Atorvastatin did not demonstrate mutagenic or clastogenic potential in 4 in vitro tests with and without metabolic activation and in 1 in vivo assay. In animal studies, atorvastatin had no effect on male or female fertility at doses up to 175 mg/kg/day and 225 mg/kg/day, respectively and was not teratogenic.
Recurrent Stroke: In the stroke prevention by aggressive reduction in cholesterol levels (SPARCL) study, the effect of atorvastatin 80 mg daily or placebo on stroke was evaluated in 4,731 patients who had a stroke or transient ischemic attack (TIA) within the preceding 6 months and no history of a CHD. Patients were male 60%, 21-92 years (average age 63 years) and had an average baseline LDL of 133 mg/dL (3.4 mmol/L). The mean LDL-C was 73 mg/dL (1.9 mmol/L) during treatment with atorvastatin and 129 mg/dL (3.3 mmol/L) during treatment with placebo. Median follow-up was 4.9 years.
Atorvastatin 80 mg reduced the risk of the primary endpoint of fatal or nonfatal stroke by 15% (HR 0.85; 95% CI, 0.72-1; p=0.05 or 0.84; 95% CI, 0.71-0.99; p=0.03 after adjustment for baseline factors) compared to placebo. All cause mortality was 9.1% (216/2,365) for atorvastatin versus 8.9% (211/2,366) for placebo.
In a post analysis, atorvastatin 80 mg reduced the incidence of ischemic stroke (218/2,365, 9.2% vs 274/2,366, 11.6%, p=0.01) and increased the incidence of hemorrhagic stroke (55/2,365, 2.3% vs 33/2,366, 1.4%, p=0.02) compared to placebo.
The risk of hemorrhagic stroke was increased in patients who entered the study with prior hemorrhagic stroke (7/45 for atorvastatin vs 2/48 for placebo; HR 4.06; 95% CI, 0.84-19.57) and the risk of ischemic stroke was similar between groups (3/45 for atorvastatin vs 2/48 for placebo; HR 1.64; 95% CI, 0.27-9.82).
The risk of hemorrhagic stroke was increased in patients who entered the study with prior lacunar infarct (20/708 for atorvastatin vs 4/701 for placebo; HR 4.99; 95% CI, 1.71-14.61), but the risk of ischemic stroke was also decreased in these patients (79/708 for atorvastatin vs 102/701 for placebo; HR 0.76; 95% CI, 0.57-1.02). It is possible that the net risk of stroke is increased in patients with prior lacunar infarct who receive atorvastatin 80 mg/day.
All cause mortality was 15.6% (7/45) for atorvastatin versus 10/4% (5/48) in the subgroup of patients with prior hemorrhagic stroke. All cause mortality was 10.9% (77/708) for atorvastatin versus 9.1% (64/701) for placebo in the subgroup of patients with prior lacunar infarct.
Pharmacokinetics: Absorption: Atorvastatin is rapidly absorbed after oral administration; maximum plasma concentrations (Cmax) occur within 1-2 hrs. Extent of absorption increases in proportion to atorvastatin dose. After oral administration, atorvastatin film-coated tablets are 95-99% bioavailable compared to the oral solution. The absolute bioavailability of atorvastatin is approximately 12% and the systemic availability of HMG-CoA reductase inhibitory activity is approximately 30%. The low systemic availability is attributed to presystemic clearance in gastrointestinal mucosa and/or hepatic first-pass metabolism.
Distribution: Mean volume of distribution of atorvastatin is approximately 381 L. Atorvastatin is ≥98% bound to plasma proteins.
Metabolism: Atorvastatin is metabolized by cytochrome P450 (CYP450) 3A4 to ortho- and parahydroxylated derivatives and various β-oxidation products. Apart from other pathways these products are further metabolized via glucuronidation. In vitro, inhibition of HMG-CoA reductase by ortho- and parahydroxylated metabolites is equivalent to that of atorvastatin. Approximately 70% of circulating inhibitory activity for HMG-CoA reductase is attributed to active metabolites.
Excretion: Atorvastatin is eliminated primarily in bile following hepatic and/or extrahepatic metabolism.
However, Avamax does not appear to undergo significant enterohepatic recirculation. Mean plasma elimination half-life (t½) of atorvastatin in humans is approximately 14 hrs. The t½ of inhibitory activity for HMG-CoA reductase is approximately 20-30 hrs due to the contribution of active metabolites.
Special Populations: Geriatric: Plasma concentrations of atorvastatin and its active metabolites are higher in healthy elderly subjects than in young adults while the lipid effects were comparable to those seen in younger patient populations.
Pediatric: Pharmacokinetic data in the pediatric population are not available.
Gender: Concentrations of atorvastatin and its active metabolites in women differ from those in men [Women: approximately 20% higher for Cmax and approximately 10% lower for area under the curve (AUC)]. These differences were of no clinical significance, resulting in no clinically significant differences in lipid effects among men and women.
Renal Insufficiency: Renal disease has no influence on the plasma concentrations or lipid effects of atorvastatin and its active metabolites.
Hepatic Insufficiency: Plasma concentrations of atorvastatin and its active metabolites are markedly increased (approximately 16-fold in Cmax and approximately 11-fold in AUC) in patients with chronic alcoholic liver disease (Childs-Pugh B).
Toxicology: Preclinical Safety Data: Atorvastatin was not carcinogenic in rats. The maximum dose used was 63-fold higher than the highest human dose (80 mg/day) on a mg/kg body-weight basis and 8- to 16-fold higher based on AUC(0-24 hr) values as determined by total inhibitory activity. In a 2-year study in mice, incidences of hepatocellular adenoma in males and hepatocellular carcinomas in females were increased at the maximum dose used, and the maximum dose used was 250-fold higher than the highest human dose on a mg/kg body-weight basis. Systemic exposure was 6- to 11-fold higher based on AUC(0-24 hr). Atorvastatin did not demonstrate mutagenic or clastogenic potential in 4 in vitro tests with and without metabolic activation and in 1 in vivo assay. In animal studies, atorvastatin had no effect on male or female fertility at doses up to 175 mg/kg/day and 225 mg/kg/day, respectively and was not teratogenic.
MedsGo Class
Dyslipidaemic Agents
Features
Dosage
80mg
Ingredients
- Atorvastatin
Packaging
Film-Coated Tablet 1's
Generic Name
Atorvastatin
Registration Number
DRP-2273-01
Classification
Prescription Drug (RX)
Reviews
No reviews found
Product Questions
Questions
